
So why is a designer interested in learning about bioinformatics?
I’ve been asked this question many times during my stay at the Wellcome Trust Genome Campus in Hinxton, where I attended the EBI Bioinformatics Summer School two weeks ago.
At Science Practice we often work with bioinformaticians helping them build expert tools. Not long ago we collaborated with the Goldman Group at the EBI and developed Sequence Bundles. Most recently we have been testing the MinION, sequencing soil metagenomes, and working with biomedical startups and companies as part of Ctrl Group. Each project requires us to sit down for a few hours or days and learn the science behind it. This learning curve is something I personally really enjoy. And because every single project we work on at Science Practice is very different from another, there is always a lot of diverse learning going on.
So my answer to the question I was asked was:
Because I’ve got a general second-hand understanding of bioinformatics, but no direct hands-on experience of how it’s done.
And we want that first-hand “done-it-myself” experience. This keeps us very close to what actually happens in the lab, in the clinic, in the field — in the very places where this insider expertise matters. That’s why I decided to spend a week in Hinxton together with 30 researchers, postdocs, and PhD candidates from around Europe to learn the science of bioinformatics in practice.

Hinxton Hall at the Wellcome Genome Campus
The course took place at the Wellcome Genome Campus which was one of the key centres involved in the seminal Human Genome Project. One of the institutes based at the Campus is EMBL European Bioinformatics Institute — the organiser of the Bioinformatics Summer School — and one of the world’s main centres for storing, curating and providing biological sequence data. The campus itself is set in rural England in vicinity of the old Hinxton Hall.
The course had a balanced mix of theory and practice. On day one Bill Pearson from the University of Virginia kicked off with a general primer in bioinformatics as an experimental science: How do we get answers to the questions that we want answered? Which questions cannot be answered by bioinformatics at all and which ones cannot be answered just yet? What are the positive and negative controls in bioinformatics? Why do we search for similarity and what are the statistics of excess similarity? And why statistically significant structure similarity can be present even in absence of statistically significant sequence similarity?
This was followed by several short tutorials on using bioinformatics resources offered by the EBI, including using the Ensembl genome browser, querying nucleotide and protein databases online, and also requesting jobs programmatically via the command line.
All that theory was an indispensable primer for launching the main activity of the Summer School on day two — a practical bioinformatics project. There were four project teams to select from:
- Genome Assembly and Annotation
- Phylogenetics
- RNA-Seq and Networks
- Structural Biology and Functional Prediction
I chose to do the Genome Assembly and Annotation project, as it had most to do with clinical genomics and genomic medicine which is in one of our areas of interest at Science Practice.
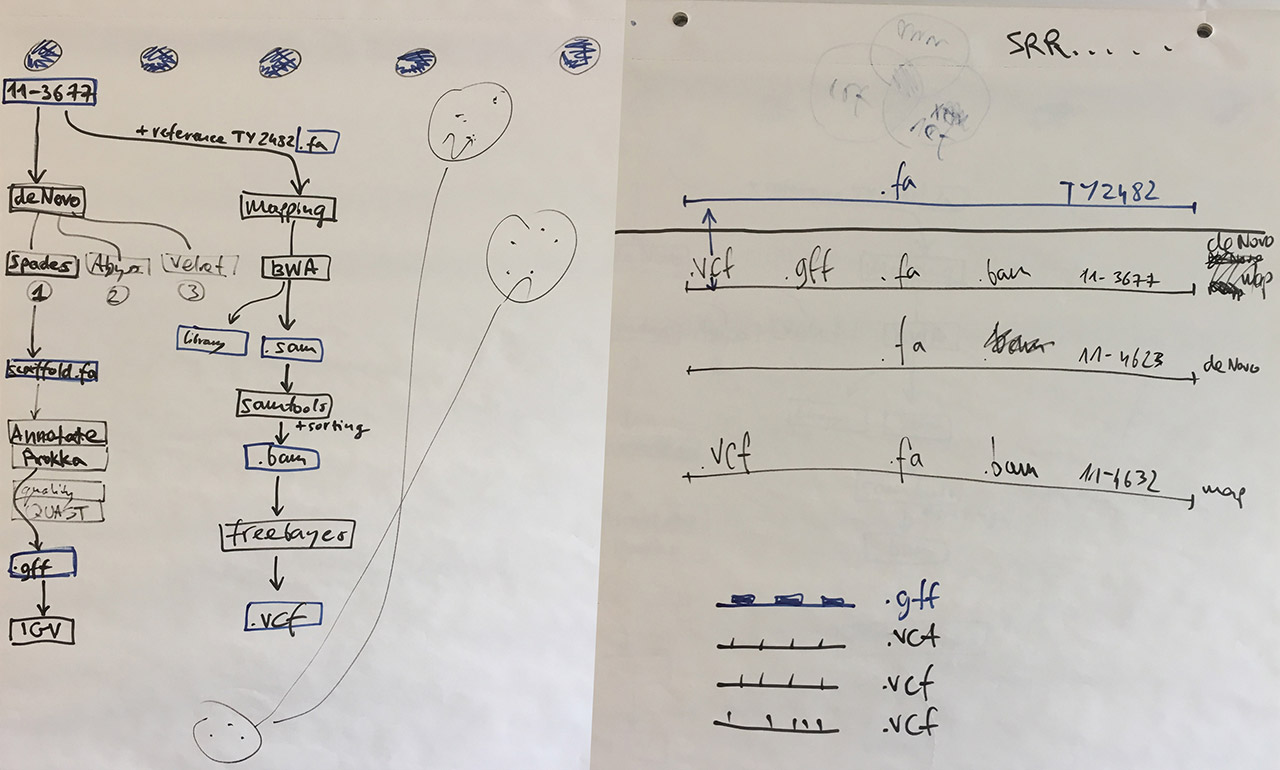
Visual notes from the bioinformatics lab
The canvas for the bioinformatics exercise was the 2011 E coli outbreak in Germany and France. The outbreak was caused by the highly pathogenic Shiga toxin-producing O104:H4 strain of Escherichia coli. Infected patients were at risk of suffering from bloody diarrhea (around 4,000 cases) which could progress to life-threatening hemolytic uremic syndrome, known as HUS (859 cases). 50 patients with severe cases of HUS died. The 2011 E coli outbreak was unusual for the rapid response in which multiplatform sequencing was brought to understand the epidemic, which is outlined in this PNAS paper.
Our task was to re-trace the investigation done during the outbreak. Working in a group of four (myself, Tracy Munthali Lunde, Massimiliano Volpe, and Dag Harald Skutlaberg) we followed these steps:
-
Obtain raw genomes from the outbreak strain sequencing experiments from the European Nucleotide Archive (data quality checked in FastQC);
-
Assemble genomes using the BWA mapping assembler — we worked with strains 11-3677, 11-4623, and 11-4632 described in the PNAS paper, and the E. coli TY-2482 reference obtained from the GigaDB database;
-
Convert, sort, index — use samtools to convert and sort assembled SAM files into an indexed binary BAM file that can be displayed in the IGV genome browser, and which can later be used for calling variants;
-
Call variants between the TY-2482 reference sequence and the assembled genomes 11-3677, 11-4623, and 11-4632 using freebayes;
-
Annotate genome using Prokka to identify which regions of the reference sequence are protein coding — this will help to understand which variants are important;
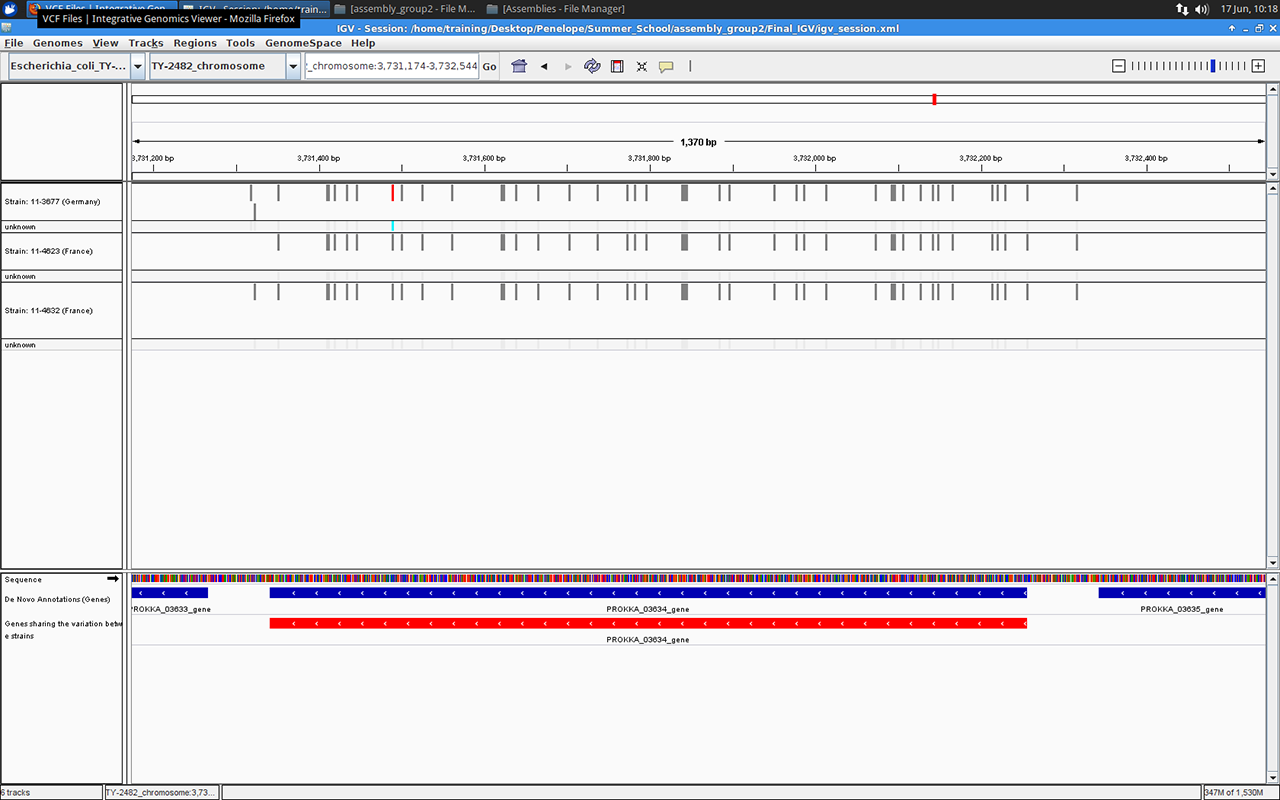
Gene number 03634 in IGV browser, screenshot by Massimiliano Volpe
We ended our project with a little investigation into the variants identified in the assembled genomes. For example we found out that about 60% of all variants in all three genomes fell within the annotated genes.
As a side project, we also managed to compare the performance of three de novo assemblers: Velvet, Abyss, and Spades. With the genome data we used, the comparison was won by Spades.
In the end, our process involved running the sequence data through a series of tools operated from the UNIX command line. Even though very simple, lacking any investigation into detailed algorithm settings and tested only on three datasets; by the end of the EBI Summer School we have succeeded to prototype our first bioinformatics pipeline.
Producing one's first ever VCF file feels great! 543 variants in a 5mbp E. coli genome. Highly toxic serotype! pic.twitter.com/dBsDNELuHQ
— Science Practice (@sciencepractice) June 15, 2016
The EBI Bioinformatics Summer School was a fantastic learning experience that will help us do even more in the prospective bioinformatics and genomics projects at Science Practice. For those interested in trying out some bioinformatics on their own, here are the Lab Notes (shared under CC-BY-4.0) that I prepared during the course. Happy assembling!